(1)Medical School and (2)Department of Computer Science, University of Minnesota, Minneapolis, MN (3)DOE Plant Research Laboratory, Michigan State University, East Lansing, MI
email: comments@lenti.med.umn.edu
The purpose of this electronic tutorial is to explain the work that we have done to date and to demonstrate methods for accessing the mountains of data. In addition, we will take you step by step through example reports, providing tips regarding our presentation of the data.
Score: 58 Frame = 2 FLRDITMVTTMVTDMD*LAISTIDSMTGLVRVFKRLLSPEFGELLKMILESLHLFFDSIS F+ + + T+ + ++ + + ++ LL + LL+ L +++L+++ +S FMLSLIYLFTLRYTLKVFSCEIFIKIVNAFALYGSLLYISYIILLNFGLLNFNLIYEHLSand sometimes the hit contains LCC regions, but still appears important:
Score: 249 Frame = 1 TSNLNYRFYDRSCPRLQTIVKSGVWRAFKDDSRIAASLLRLHFHDCFVNGCDGSILLNDS +++L+ RFY RSCPR Q+I++ GV A + + R++ASLLRLHFHDCFV+GCD+SILL+D+ SAQLSPRFYARSCPRAQAIIRRGVAAAVRSERRMGASLLRLHFHDCFVQGCDASILLSDT
In both of the above examples, lysine occurs much more frequently than would be expected for a random distribution, where there would be approximately one lysine in every 20 residues.
A few other items are taken into consideration in processing the sequences. When DNA libraries are made using directional cloning, only the positive strand is analyzed. If the clones are not directionally cloned, as is the case for some of our other projects, then all six frames are processed. The blasts are performed against current versions of GenBank, PIR and GenPept databases using a PAM score (Dayhoff et al., 1978) of 250. The high PAM score has been chosen because it allows for similarity matches, or hits, between sequences of large evolutionary distance, and plant DNA makes up a relatively small proportion of the public databases. This high PAM value does not affect matches to very similar sequences (i.e., other plants). Finally, the EST sequences are re-blasted on a reasonably regular basis to keep up with additions to the public databases.
As another step in making data accessible, we submit all sequences that are at least 200 base pairs in length with no more than 5% unknown bases (these criteria were defined experimentally, Shoop, et al., 1994) to dbEST (Boguski et al., 1993) at NCBI. The individual clones, which contain the EST sequences, are also made available at the ABRC Stock Center at Ohio State University.
After searching, the server will provide you with a list of documents appearing as links (Figure 2). Each of these documents contain all the information described above (Section 2.0) for a single cDNA, and only the documents containing the key word(s) for which you searched are listed. If any the fields that you selected have no hits, this is reported at the top of this list. For example, when searching the MSU Arabidopsis sequences and the USDA loblolly pine sequences for "catalase", we find that there are no hits for the pine sequences; therefore, the hits listed are all from the Arabidopsis sequences (Figure 2). The default value for the number of documents returned is 200. If you get 200, it is probably because there are a lot more than 200 documents that contain your search words. This list of documents is ordered based on the score that the freeWAIS program, which is used to make the WAIS index, assigns to each document. This score is based on the number of occurrences of the search words in a document, the location of the words in a document, the frequency of those words within the collection, and the size of the document. It is important to keep this in mind as you survey your results, because, for example, a high score may point to a document that contains very few or no data.
http://lenti.med.umn.edu/r2.MSU_Ath/950315b/154N18T7-JCAG500.seq.html
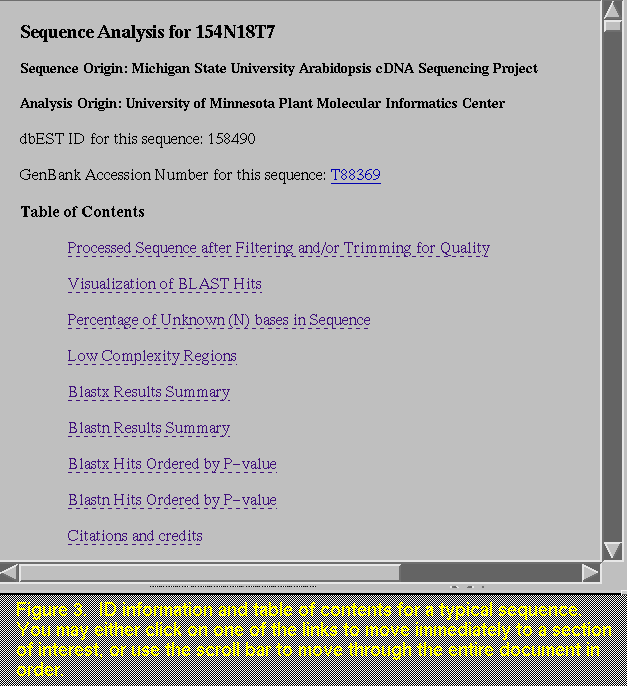
that is seen in Figure 2, the file containing the sequence analysis information for clone 154N18T7 is opened (Figure 3). This document can be very large, and it is generally advantageous to review its contents by scrolling through various parts of the document and clicking on links to hop quickly to other parts. Another way to move to items of interest is to use the "Find In Current" (or "Find") feature under the "File" (or "Edit") menu, which is part of Mosaic (or Netscape) itself. Here you can type in a key word, such as what you used for the WAIS search, and find the location of the words in the current document. For now, we will use the scroll bar on the right side of the file to take you through the document. First, we move directly to information about the EST studied in this file (Figure 4). The DNA sequence is presented at the top of this file so that you may copy it for your own records.
Following the background sequence information, a visualization of BLASTX similarity results is presented with an image from the Alignment Viewer tool (Figure 5), followed by a visualization of BLASTP hits. In these images, all the alignments to the sequence in question are plotted according to score on the y axis and according to the location and length of the alignment on the x axis. Alignments with scores of 150 or greater are considered strong hits, and a putative function can be assigned ESTs which have hits with this high a score (Shoop et al., 1994). Alignments with scores of less than 80 should be viewed critically, as such a low score suggests that the alignment was largely due to chance. Also, short regions of high similarity may have an artificially high score.
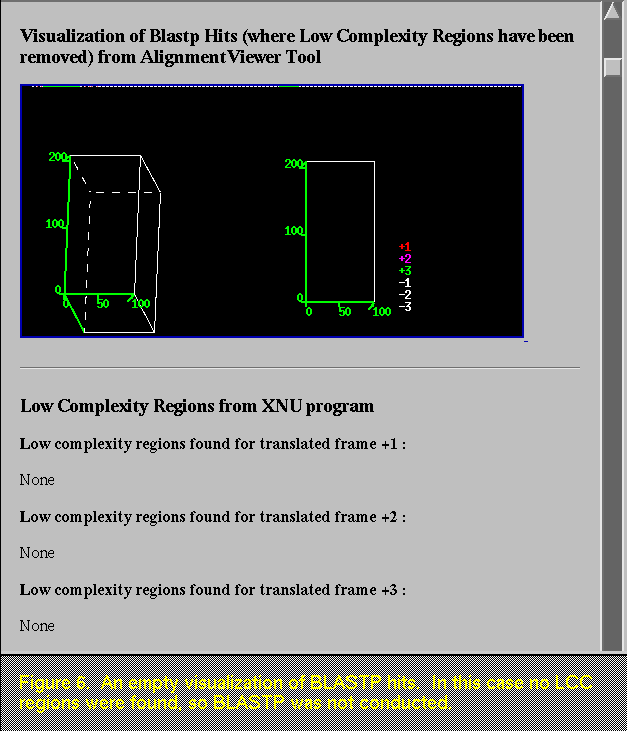
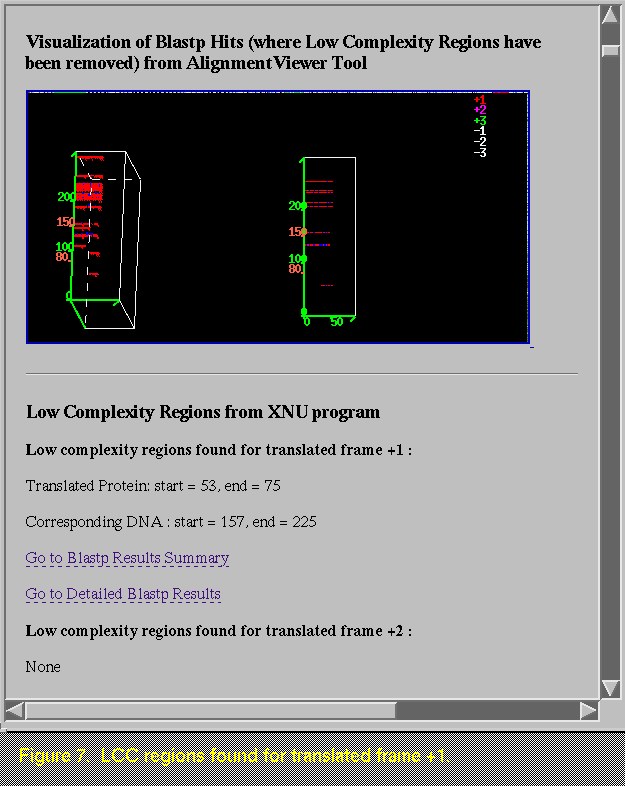
Sometimes the BLASTP image appears empty (Figure 6), and this can occur for various reasons. For instance, if there are no LCC regions found in the sequence, the BLASTP program is not run. As another example, there may be LCC regions found in frame 1, but alignments only in frame 3. In this case, a BLASTP was run on frame 1, but since there are no hits to frame 1, there are no data to graph. If there are LCC regions found in a frame that shows sequence similarity to a public sequence, the location of the regions is presented, as shown in Figure 7.
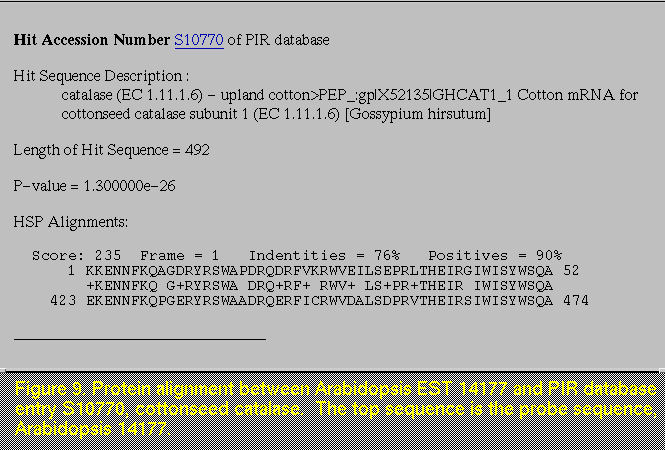
Underneath the description of LCC regions, a link allows you to go directly to a summary list of protein alignments that were obtained with BLASTP (Figure 8). This summary enables you to determine at a glance what frame the alignments occurred in, whether or not there was a frame shift, and a brief description of the sequence to which the alignment occurred. Additionally, the higher scores and lower p-values indicate which hits are the strongest. By clicking on the "goto" link, you can see the actual alignment between the protein sequences, as well as a longer description of the sequence from the databank (Figure 9). You can also get to these alignments by continuing to scroll down the document from the summary list. Note that the top sequence is the test (or probe) sequence that blast was run against and the bottom sequence is the similar sequence from the target public databank. If you are interested in researching the latter sequence, there are links, which connect directly to the databank, in both the summary report and the individual alignment reports.
If, instead of using the link to go to the BLASTP data, you continue to scroll down the report, there is a summary list of protein alignments that were obtained with BLASTX. These data are collected without masking out any low complexity regions, but the presentation of the data is identical to the BLASTP data. In cases where LCC regions are found, it is important to compare the BLASTX and BLASTP results to determine which data are appropriate for your research purposes.
The summary list of BLASTX alignments is followed by the summary list of BLASTN alignments, which provides similar information on the DNA alignments obtained with BLASTN. After the summary lists are the detailed lists of BLASTX, BLASTN, and BLASTP hits (Figure 9), respectively. These lists are ordered according to increasing p-value, and hits with p-values greater than 0.1 are omitted from the list. For some ESTs, when you look for the BLASTP hits, you may find the heading of this section, but no entries (Figure 10). This may seem puzzling at first. However, if there are no low complexity regions, or hits occurred with a p-value greater than 0.1, there are no reportable BLASTP results, and hence, this section should be empty. If you go back to the table of contents at the top of the document, and click on the "Low Complexity Regions" title, you can double check whether or not LCC regions were found.
First, we have a local BLASTN search tool ready, and it will likely be on our server before you read this paper. This will allow you to search our data sets with your DNA sequence(s). It is interactive, and web-based, and all you need do is select the target genomes you are interested in, paste your sequence into the window, and click on the "submit your query" button. An obvious follow-up for this program is a version for BLASTX to run your data against the translations of coding sequences in our data. All we are waiting for to bring this up is the new version of Xgrail (Xgrail 1.3; unfortunately, the current version isn't suitable for our needs) from Oak Ridge National Labs so we can have a smarter version of the translations of the sequences.
Second, Alignment Viewer (AV) (Chi et al., 1995) is the tool we use to produce the static images in the EST analysis reports. In its "real" incarnation, the program is 3-dimensional and interactive, and additional information is calculated and displayed "on the fly" (specifically, it shows you a curve of what the substitution matrices will do if you were to run them all; both PAM (Dayhoff et al., 1978) and BLOSUM (Henikoff and Henikoff, 1993) matrices are included. If you have a Sun (SunOS 4.1.3, Solaris 2.4), SGI or Linux computer running the "motif" window system, you will be able to run this client. It functions much like other external viewers for the Web (xv being one of them for Unix-based machines), in that it resides on your machine, but reads data from ours.
Third, the MotifExplorer (Bieganski, et al., 1996) is based on a something called a "suffix tree" (Bieganski, et al., 1994; Bieganski, 1995). The underlying algorithm and data structure is described in the papers above; what it *is*, however, is a very fast pattern matching tool. This is the only large scale implemetation that we know of, and presently allows you to explore PIR using a variety of patterns, including ProSite patterns or those of your own design. Our own data will be available under this tool, shortly after we get Xgrail 1.3 (see note above).
One of our longest term and most complex projects has been the development of a relational database management system (DBMS) for sequences, lab, analytical and derived information. This system will allow for very complex searches, or queries, against the entire data set. The data structure has been defined, and, indeed, data has been loaded into the database. It is still very much under development, and is queriable only by Structured Query Language (SQL) at the moment; however, we are designing web-based "query-building" screens so that one will not have to understand SQL (or even know that it exists) to use the DBMS. One of the unique additions to this system has been the development of database operators (termed "datablades") for the DBMS that understands protein motifs (Lundberg, 1995). This allows the searching of sequences in the database with any pattern and particularly the ProSite patterns.
As happy as we are with the directions we have taken for this project, EST data, even extended by a variety of analyses, needs a context to exist in. To that end, we have been volunteering our time to work on other genomes, feeling that some of the real treasures may be in the comparative genome analysis. While this part of the project is presently unfunded, we are gathering ESTs from rice, maize and loblolly pine presently, and we will shortly add some of the Brassica sequences. In addition, some preliminary discussions are in progress with other projects. We feel very strongly that this database of multi-genus plant ESTs has the ability to begin to provide direction both for molecular and for biological experimentation.
We would also like to give our thanks to Mary Anderson of the Nottingham Arabidopsis Stock Centre, and Carolyn Tolstoshev, Mark Boguski and Jane Weisemann of NCBI's dbEST; their encouragement and assistance in this work has been very important to us.
P. Bieganski. 1995. "Genetic Sequence Data Retrieval and
Manipulation based on Generalized Suffix Trees." Ph.D. Thesis,
University of Minnesota, Minneapolis, MN.
P. Bieganski, J. Riedl, J.V. Carlis and
E.F. Retzel. 1994. "Generalized Suffix Trees for Biological
Sequence Data: Applications and Implementation." In: Proceedings
of the IEEE 27th Hawaii International Conference on System
Sciences. Oahu, Hawaii. L. Shriver and L. Hunter, (Eds.). IEEE
Computer Society Press. V:35-44.
P. Bieganski,
J. Riedl, J.V. Carlis and E.F. Retzel. 1996. "Motif Explorer--A Tool for
Interactive Exploration of Amino Acid Sequence Motifs." Pacific
Symposium on Biocomputing, Hawaii. Submitted.
M.S. Boguski, T.M.J. Lowe, and
C.M. Tolstoshev. 1993. "dbest - database for expressed sequence
tags." Nature Genetics, 4:332-333.
Ed Huai-hsin
Chi, Phillip Barry, Elizabeth Shoop, John V. Carlis, Ernest Retzel,
John Riedl. 1995. "Visualization of Biological Sequence
Similarity Search Results" Accepted for "IEEE Visualization
'95" October Conference. Atlanta.
Jean-Michel Claverie and David
States. 1993. "Information enhancement methods for large scale
sequence analysis." Computers and Chemistry,
17(2):191-201.
M. O. Dayhoff, R. M. Schwartz, and
B. C. Orcutt. 1978. "A model of evolutionary change in
proteins." In: Atlas of Protein Sequence and Structure,
M. O. Dayhoff, (Ed.). National Biomedical Research Foundation, Vol. 5,
Suppl. 3, chapter 22, 345-352.
Warren Gish and David
States. 1993. "Identification of protein coding regions by
database similarity search." Nature Genetics,
3:266-272.
Steven Henikoff and Jorga Henikoff. 1993. "Performance
evaluation of amino acid substitution matrices." Proteins: Structure, Function, and
Genetics, 17:49-61.
Ann M. Lundberg. 1995. "Extension of a DBMS with Protein
Motif Search Capabilities." M.S. Thesis,
University of Minnesota, Minneapolis, MN.
T. Newman, F. de Bruijn, P. Green, K. Keegstra, H. Kende, L.
McIntosh, J. Ohlrogge, N. Raikhel, S. Somerville, M. Thomashow, E.F. Retzel and C.
Somerville. 1994. "Genes Galore: A Summary of Methods for Accessing Results from
Large-Scale P artial Sequencing of Anonymous Arabidopsis cDNA
Clones." Plant Physiology. 106:1241-1255.
E. Shoop, E. Chi, J.V. Carlis,
P. Bieganski, J. Riedl, N. Dalton, T. Newman and
E.F. Retzel. 1995. "Implementation and Testing of an Automated EST
Processing and Similarity Ana lysis System." In: Proceedings of
the IEEE 28th Annual International Conference on System
Sciences. Maui, Hawaii. L. Shriver and L. Hunter, (Eds.). IEEE
Computer Society Press. 5:52-61.
E. Shoop,
J.V. Carlis and E.F. Retzel. 1994. "Automating and Streamlining
Inference of Function of ESTs within a Data Analysis System" In:
Proceedings of the IEEE 27th Hawaii International Conference on System
Sciences. Oahu, Hawaii. L. Shriver and L. Hunter, (Eds.). IEEE
Computer Society Press. V:45-46.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}