G[ACG]GATC>
locates over 3,700 fragments matching the patterns GAGATC, GCGATC, and
GGGATC, but the report indicates that only three examples from the latter two patterns are found
at the 3' end of the sequence.
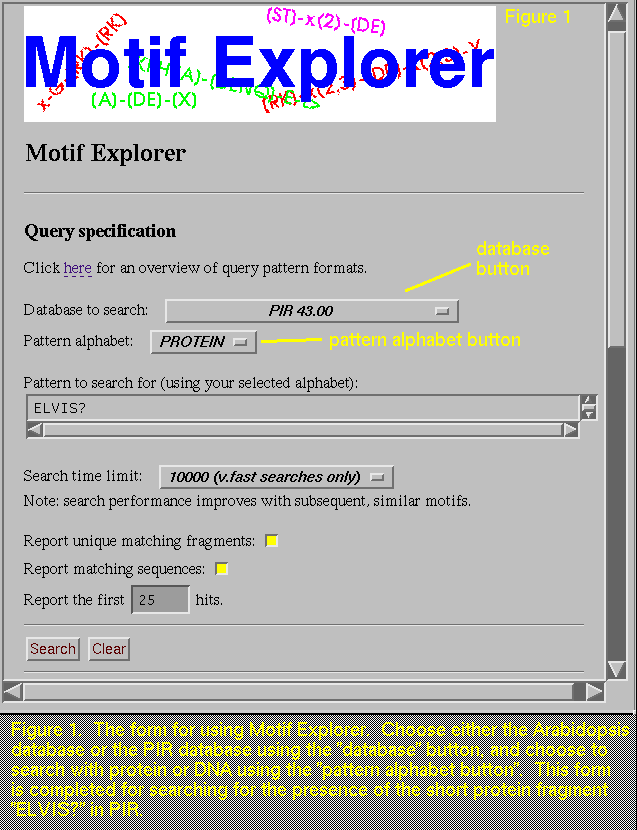
There is a slight oddity you may notice
when you run a search using the left pointy bracket,
"<". This symbol is used by Motif Explorer to designate
the beginning of a sequence, and it is also has meaning for HTML (the
language we use to make Motif Explorer available to you on the
server). Due to this double use of "<", the "Query
expression" line on the report will show up blank, but this is
not cause for concern, as the search and its results are not
affected.
Additional searching power is gained using regular expressions, a
pattern searching technique that is commonly used in computer
science. The ? (question mark) character, the | (vertical bar, or "pipe")
operator, and parenthesis are used along with the letters of the DNA
(or protein) alphabet to make these expressions. The ? (question mark)
character allows for any base (or amino acid) to be accepted, just as
with the "x" in PROSITE conventions, and the | (vertical
bar, or "pipe") operator acts as an "or statement". For example, if you
were looking for EcoRI or AlwnI restriction enzyme digestion sites,
you could enter the following search:
(GAATTC)|(CAG???CTG)
to find matches to GAATTC, or any string of bases that starts with
CAG, ends with CTG and has any 3 bases in between. Parenthesis can
also be used to nest the expressions. By searching on AC((AG)|(CC))T,
you will match sequences ACAGT and ACCCT.
The more complex queries may require that you change some of the other
parameters on the form. Many searches will get completed using the
default "very fast" search time. Sometimes your search will
take longer, and the 20 second time limit is appropriate. Chances are,
if you need to conduct a search that has no time limit, the results you
obtain are likely not to be useful to your work. If you
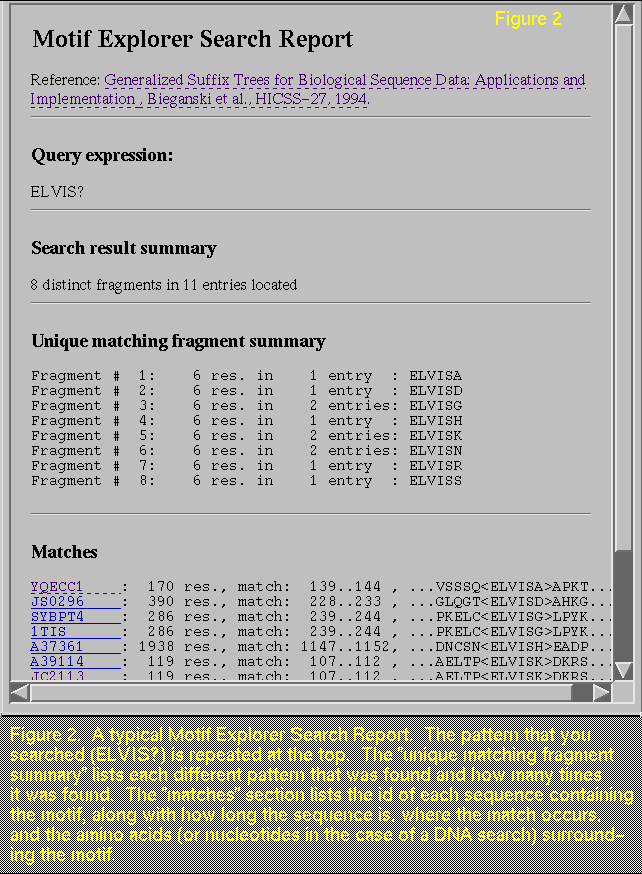
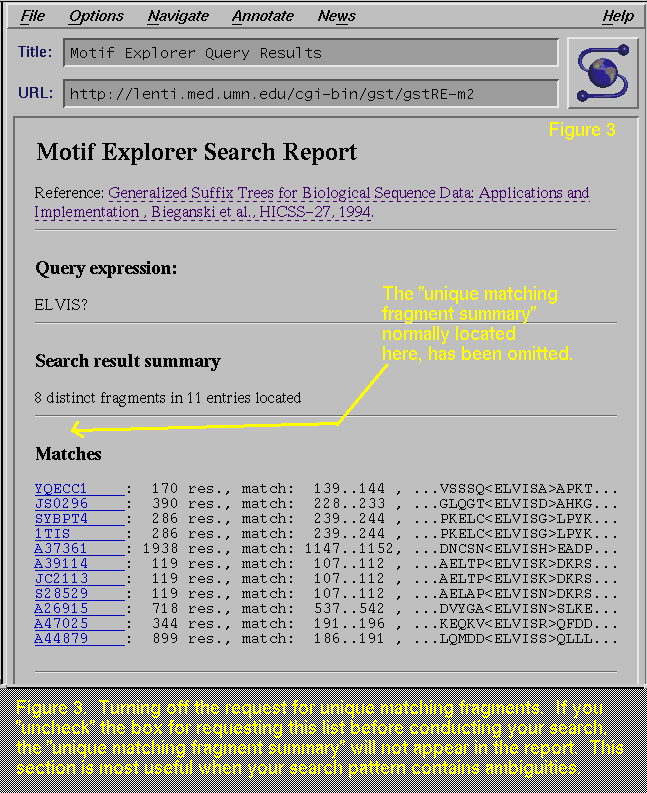
"uncheck", or turn off, the "Report unique matching
fragments" box, then the report will not show the "Unique
matching fragment summary" section, which is missing in Figure 3 (compare with Figure 2, which has this section). This section is most
useful when your search may find more than one distinct pattern.
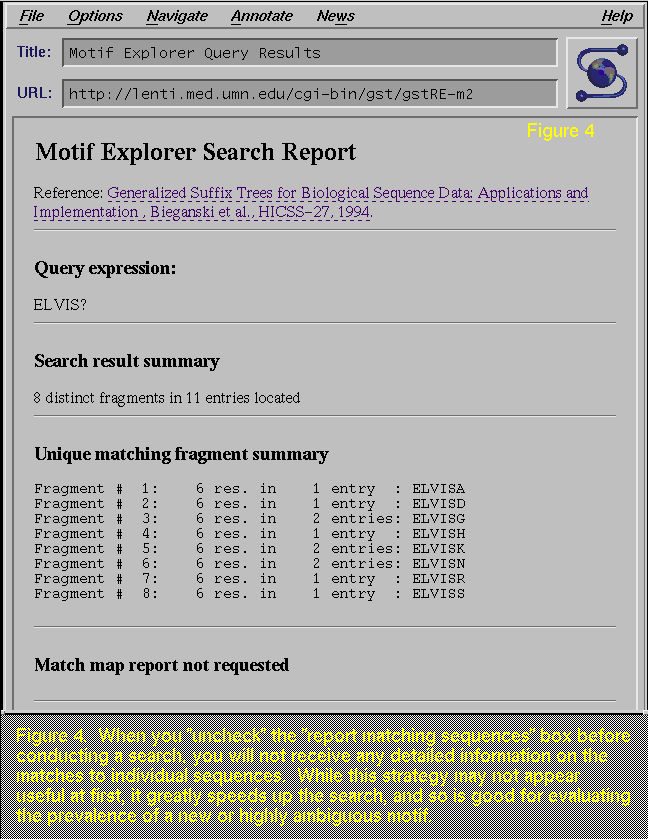
By
"unchecking" the "Report matching sequences", you
will not get the list of matches (Figure 4; compare with Figure 2). While this step
drastically limits the amount of information you will receive in the
report, it does speed up the query time, and is useful when you are
testing out the prevalence of your search pattern.
Currently, if you conduct a protein search on the MSU
Arabidopsis DNA database, the characters are interpreted as DNA
bases, not as amino acids. That is, when you input a peptide
sequence, it is back-translated, using full redundancy, to the appropriate nucleotide
sequence(s) before running the search. (Similarly, motifs for DNA
searches on the PIR database are translated to protein before the
search is conducted.) In the future, we plan to perform reading frame
prediction using GRAIL to reduce the likelihood of false-positive
hits. Arabidopsis sequences will then be translated into
probable reading frames and searched as polypeptides when you conduct
a search using a protein motif. Also, we will eventually be putting
all Arabidopsis sequences available at NCBI's dbEST in the
Motif Explorer Arabidopsis DNA database; presently, as mentioned above,
only those from the
MSU project are included.
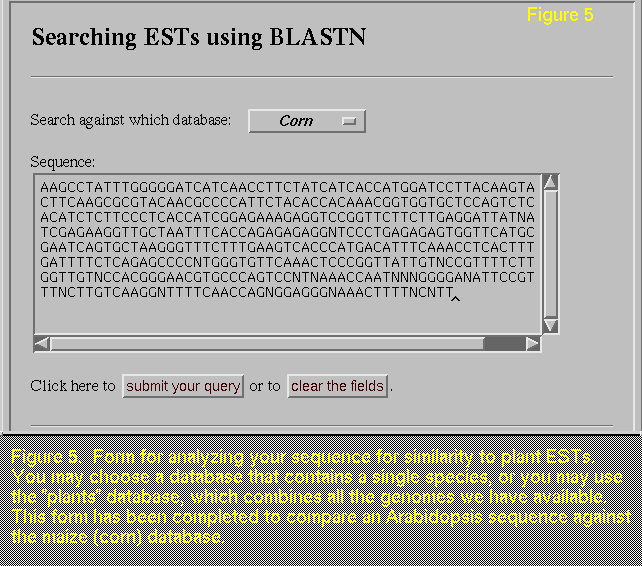
If you have sequenced a gene from Arabidopsis or another plant,
you will be interested in the plant EST databases that we have created
by compiling Arabidopsis, maize, rice, and loblolly pine
sequences. Using the form provided (a completed example is shown in
(
{kind=link}
{kind=link}
{kind=link}